
Post-market surveillance is already an important component of human factors analyses that inform the development of any safe and effective medical device. For manufacturers of Software as a Medical Device (SaMD) that elect to participate in the FDA’s Digital Health Software Precertification Program, post-market surveillance data will become even more important. SaMD in many cases presents opportunities for a manufacturer to monitor Real World Perfomance Data (RWPD) directly via data recorded by the SaMD itself, but analysis of the events catalogued in publicly available adverse event databases remains a key source of post-market surveillance. The FDA’s MAUDE database can be complicated and time-consuming to mine. To make the most of this data, manufacturers would benefit from having a more efficient means by which to extract meaningful insights.
Background
By implementing the pre-certification program, the CDRH at FDA hopes to establish a regulatory model that will “assess the safety and effectiveness of software technologies without inhibiting patient access to these technologies.” Under the FDA’s program, manufacturers of SaMD who successfully obtain precertification can benefit from streamlined premarket review, and, in cases of some lower-risk software or certain types of modifications, can bypass FDA review all together.
In order to allow for streamlined (or sometimes bypassed) premarket review, but still achieve its mandate to ensure the safety and efficacy of medical devices, the FDA must verify through the pre-certification program that a manufacturer has established a “robust culture of quality and organizational excellence” and is “committed to monitoring real-world performance of their products once they reach the U.S. market.” In their working model of the pre-cert program, the FDA outlines the key components to the program (Figure below taken from Developing a Software Precertification Program: A Working Model).
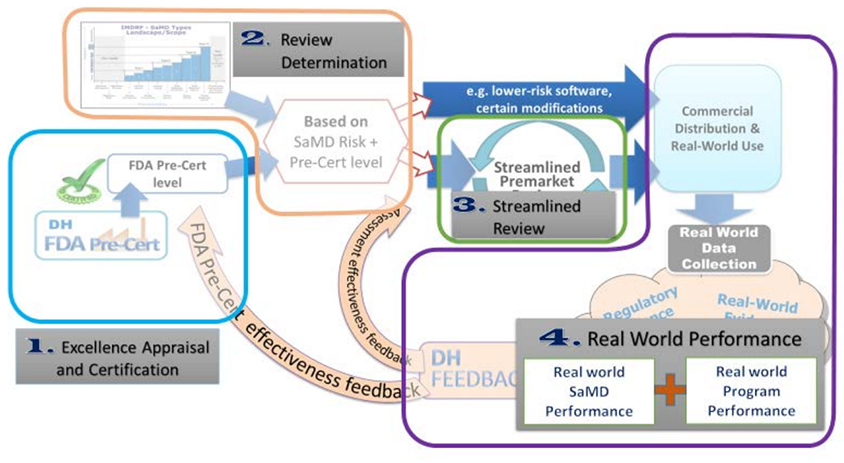
From a human factors perspective (and likely from other perspectives), the details of the excellence appraisal and certification are critical to the value and the success of the program itself. The FDA is working with the pilot participants in the program to define those details, which will be centered around five key principles:
- Product Quality
- Patient Safety
- Clinical Responsibility
- Cybersecurity Responsibility
- Proactive Culture
One can hope that sound human factors practices (e.g., documented institutionalization of getting feedback from end users early and often, and grounding design decisions in this feedback) are threaded throughout the assessment of an organization against these principles.
Post-market surveillance data is key
Real World Performance Data (RWPD, i.e., post-market surveillance data), and the extent to which regular monitoring of this data to is relied upon to inform continuous improvement, is an important component of the pre-certification program. With a streamlined (or bypassed) regulatory review, implementing a robust process by which RWPD is constantly monitored, analyzed, and acted upon will be critical to manufacturers’ demonstration to the FDA that their organization is committed to producing SaMD that is both safe and effective.
RWPD includes three key components in and of itself:
- Real World Health Data (RWHD)
- User Experience Data (UXD)
- Product Performance Data (PPD)
In many cases, software by its very nature presents opportunities for monitoring RWPD that require minimal effort to at least obtain the data on the part of the manufacturer. For example, error reports that are automatically sent to a manufacturer from a software program can be monitored for reliability analysis. In other cases, where data are (at least for now) generated externally, such as is the case with adverse event and product complaint reporting, more effort is required to obtain RWPD.
The data are made publicly available in one way or another – whether formally via databases such as the FDA MAUDE or FAERS databases, or informally via other channels such as social media. In both cases, independent of the effort required to obtain the data, a manufacturer of SaMD seeking precertification through this program must have a robust process in place to collect, analyze, and act on this data; any medical device manufacturer, for that matter, should also have this process in place. As is increasingly the case in the information age, the problem becomes not how to obtain data, but how to manage the incredible amount of data available.
Making post-market surveillance data easier to consume
While there are multiple sources for post-market surveillance data, the MAUDE database remains the more relevant “official” database of publicly available AE/PC data relevant for SaMD manufacturers. Unfortunately, the MAUDE database is not very accessible and requires a database engineer to plumb the depths of the data before any meaningful analysis can be conducted.
That said, with the right skills brought to bear, there is a wealth of information available in MAUDE for SaMD manufacturers to leverage in their efforts to ground their designs in RWPD. This seems to be an opportunity for a subscription service that regularly summarizes trends in AE/PC data from the MAUDE database and other sources. The MAUDE database in particular is not without its limitations, several of which are stated very clearly on the FDA website itself, but as long as it is the official database of AE/PC data for medical devices it would seem beneficial to have easy access to emerging and historical trends in this data. This way, medical device manufacturers in general could be better able to institutionalize the incorporation of robust post-market surveillance data analysis into their design process, and SaMD manufacturers in particular could better prepare themselves for precertification and streamlined regulatory approval process in doing so.